

サブマリン・ゲル電気泳動を行う際の問題は,サンプル溝に試料を入れるのをゲルを緩衝液に入れる前・後いずれに行っても,入れた試料が液中に出てしまいやすいこと。したがって,試料のロ-ドには十分に注意が必要である。
ただし,ゲルの種類によっては,より低濃度にすることもできる。製品名「シ-プレップ」。
溶けたゲルをゲル作成用の入れ物に注ぐ際は,ゲルの温度が60゜C前後に下がってからにする。あまり熱いと入れ物が熱で変形する可能性がある。(入れ物は通常,プラスチックでできていることが多い)ゲルを注ぐと同時にサンプルコ-ムを入れる。やがて,ゲルが十分に個化した後,静かにコ-ムを抜く。
TAE 緩衝液 の原液は, トリス 242 g
氷酢酸 57.1 g
0.5M EDTA 100 ml
これを水1リットルに溶かすとpH8.0になる。
この原液を50倍に希釈して使用する。
詳しくは,別紙を見よ。
5M Urea
10%グリセロ-ル
0.025%BPB
0.5%SDS
<泳動条件>
泳動は,「ミュ-ピッド」を用いた場合,約50mV前後で行う。
アガロースゲルの作成・サンプルのウェルへの注入
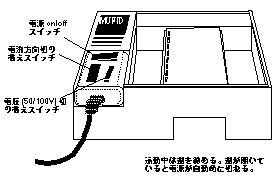

電気泳動装置・ゲルの染色
【ゲルの染色法】
一般に,DNAの染色にはエチジウムブロマイドが用いられる。しかし,この染料は高価なうえに発癌性がある。それに対して,著者は,アズ-ルCという従来細胞学の分野でよく用いられてきた染料を用いている。これはおそらくDNAのリン酸基に結合すると思われる。したがって,エチジウムブロマイドのように泳動のDNAを染色して見ることはできない。しかし,染色感度や染色の手軽さ,その他の扱いは同じである。(感度については調べた限りでは, ugの量を検出できる)。なによりも使用上の危険性がないのが大きな特徴といえる。
アズ-ルC 今使用中のものは,
東京化成工業(株) 製造のもの。
ロット番号は,A396 である。
pHは,中性の条件でもっともよく染まるので,pH7.0のリン酸緩衝液を少々(10mM前後か)加えておく。
【染色後のゲルについて】
1)保存方法
アガロ-スゲルは,これをゲルボンド(でなくともよい。なにかゲルが乾燥した際に,密着できる親水性のあるもの)などの支持板の上に置いて自然乾燥させる。脱色が適当な場合,きれいなまま乾燥,保存ができる。しかし,今のところまだ,どうすれば適当な脱色かを判定できる規準がみつかっていない。あまり脱色しすぎれば,バンドそのものが消えてしまう。バックグラウンドが気にならないくらい脱色されかつ核酸のバンドが十分染色されている状態であればいつでもきれいなまま乾燥できるかというとそうでもなく,乾燥するにつれてわずかにのこっていた染料が局部的に集まりきたなくなる場合も多い。
2)ゲルから核酸を回収する方法 Electroelution
これは,いわゆる「エレクトロエル-ション」といわれるものだが,アズ-ルCを使った場合,エチジウムブロマイドのように紫外線を当てないとどこにバンドがあるかわからないのと違い,肉眼でバンドの位置が確認できるのが特徴である。したがって,そのまま青く染まったバンドの部分を切り出し,それらを透析チュ-ブにいれて,おなじ泳動槽の中で泳動するのである。すると,それまで結合していた核酸とアズ-ルは,核酸がプラス極側,アズ-ルがマイナス極側に移動し,分子量の小さいアズ-ルは,透析チュ-ブを通り抜けるが,核酸は分子量が大きいので,チュ-ブ内に残る。
その後は,チュ-ブから,核酸の混じった液を取り出せばいいのだが,実際は,ゲルの細かな破片が混じってくる。これらは,以後の酵素処理などに影響する場合があるので,除いておく必要がある。また,核酸の濃度もかなり希釈されているので,濃縮するにも,通常のエタノ-ル沈澱では,ロスが多。そこで,いったんブチルアルコ-ルと混ぜてはげしく振る。これを遠心すると,水に比べて思いブチルアルコ-ルは下相にくるので,水相を取り,再びブチルアルコ-ルをこれに加える。このように,何度かアルコ-ルを交換すると,そのあいだに水分が一定の割合でブチルアルコ-ル側に吸収されるので,核酸が濃縮される。かつ,その間の遠心によって固形分のアガロ-スゲルの混入物を除去することもできる。
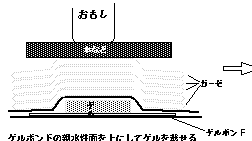

ゲルからの脱水・ゲルボンド上での乾燥
◆ ゲルからのDNAの回収(DE81法)
1)ゲルのスリットの巾に合わせて,DEAE-Cellulose paperを切る。
2)2.5M NaCl に一晩浸し,TA buffer で3回洗う。
3)ゲルを染色し,カッターで切り込みを入れ,paper をはさみ込む。
(レーン間を開けておくとよい。)
4)さらに,泳動する。(吸着)
5)吸着したpaper をeppendorf tubeに入れ,DE81法溶出液* を300ml 入れる。
* 20mM Tris-HCl (pH 7.5)
1 mM EDTA
1.5M NaCl
6)paper をvortexや,ピペットで断片化する。
7)37゚C incubate 3 hr ~ over night
8)遠心 12,000 rpm, 5min x 2
9)上澄みをピペットマンで回収
10)water saturated n-butanol 1 ml を加え EtBr を取り除く。
(上層が n-butanol)
11)Carrier (tRNA 1ul, 10 ug )を加える。
12)エタノール沈澱をする。
13)75%エタノールで洗う。
*これは,遠藤君から教えてもらった方法。ゲルに直接,DEAEセルロース紙をはさんでそこにDNAを吸着させるという簡便法だ。
しかし,簡便法とはいえ時間がかかり過ぎるような気もするが。